




Informaţii generale
Anomalii cromozomiale numerice
Cariotipul unei specii, termen introdus de Levitsky in 1924, reprezintă numărul de cromozomi prezenţi în nucleul celulelor somatice. Cariotipul uman este alcătuit din 46 cromozomi, organizaţi în 23 perechi; prin analiza citogenetică, cromozomii pot fi identificaţi ca entităţi structurale discrete, fiecare celulă umană conţinând 22 perechi de autozomi, precum şi o pereche de cromozomi sexuali. Intr-o pereche de cromozomi unul este de provenienţă maternă, iar celelălalt de origine paternă1.
Un set de 23 constuie numărul haploid (N) de cromozomi şi corespunde numărului de cromozomi dintr-un gamet. In procesul de fertilizare două seturi haploide se alătură pentru a forma zigotul cu 46 cromozomi – setul diploid (2N) de cromozomi. Erorile survenite în diviziunea celulară pot genera seturi de cromozomi care au mai mult sau mai puţin de 46 cromozomi. Prezenţa într-o celulă somatică a unui multiplu exact al unui set haploid de cromozomi este denumită euploidie, în timp ce pierderea sau prezenţa în plus a unuia sau mai multor cromozomi este cunoscută ca aneuploidie.
Diploidia – starea normală a celulelor umane – reprezintă o forma de euploidie (eu – “good”, ploid – “set”) . Poliploidiile anormale includ triploidia (3N = 69 cromozomi) şi tetraploidia (4N = 92 cromozomi), aberaţii ce nu sunt compatibile cu viaţa şi care sunt depistate în principal în produşii de concepţie avortaţi spontan. Triploidia se poate datora unui eşec în gametogeneza apărut la una din diviziunile meiotice, ce dă naştere unui gamet 2N, care atunci când este fertilizat de către un gamet haploid provenit de la celălalt părinte, va produce un zigot triploid. Pe de altă parte un set 3N mai poate fi derivat din dispermie – fertilizarea unui ou haploid de către 2 spermatozoizi – ce dă naştere de obicei la o mola hidatiformă parţială. Tetraploidia este în majoritatea cazurilor un eveniment postmeiotic care se prezintă ca o duplicare a unui set diploid (XXXX sau XXYY), ca urmare a unui eşec apărut într-un clivaj mitotic precoce al zigotului1;3.
Aneuploidia are ca mecanism de producere nondisjuncţia (lipsa de separare a comozomilor), care poate surveni fie în meioză, fie în mitoză. De obicei este afectată o singură pereche de cromozomi. Nondisjuncţia mitotică precoce într-un zigot poate da naştere la o aberaţie cromozomială prezentă în toate celulele din organism, în timp ce erorile de diviziune survenite mai tarziu conduc la mozaicism – prezenţa în acelaşi organism a două linii celulare ce diferă între ele printr-un cromozom.
In meioza normală o replicare ADN este urmată de două diviziuni celulare ce au ca rezultat final formarea de gameţi haploizi. Prima diviziune meiotica se caracterizează prin reducerea la jumătate a numărului total de cromozomi pe celula. A doua diviziune meiotică este o simplă diviziune de tip mitotic în care are loc separarea centromerilor şi distribuţia cromozomilor în celulele-fiica. Erorile survenite în meioză pot da naştere la gameţi cu un cromozom în deficit sau în exces. Nondisjuncţia din cursul primei meioze va genera 2 gameţi care sunt disomici pentru un cromozom şi la 2 gameţi cărora le lipseşte cromozomul respectiv (nulisomici). Atunci când are loc fertilizarea, primii gameţi vor da naştere la un produs de concepţie cu trisomie, în timp ce în al doilea caz va rezulta un zigot cu monosomie. Atunci când eroarea survine în cea de a doua diviziune meiotică rezultă doi gameţi haploizi normal, un gamet disomic şi unul nulisomic (vezi fig.19.1.1).
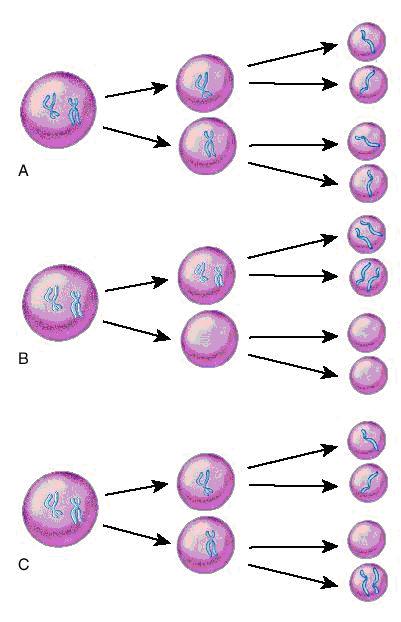
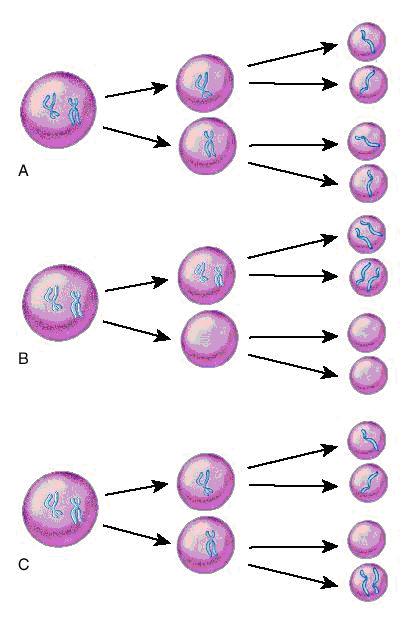
Fig. 19.1.1 Meioza
A. Meioza normală; B. Nondisjuncţie în prima diviziune meiotică; C. Nondisjuncţie în a doua diviziune meiotică
(Adaptare după medical-dictionary.thefreedictionary.com)
Trisomia şi monosomia pot afecta orice cromozom, însă majoritatea acestor aberaţii sunt incompatibile cu viaţa şi vor da naştere la un avort spontan. De exemplu, trisomia 16 este anomalia cea mai frecvent raportată la analiza citogenetică a produşilor de concepţie avortaţi insă ea nu a fost descrisă la nou-născuţii vii. Trisomiile autozomale care se pot însoţi de un făt viabil sunt cele care includ cromozomii 13, 18 şi 21. Mai rar sunt identificate persoane cu mozaicism cromozomial care conţin trisomiile 8, 9 sau 22. Trisomiile cromozomilor sexuali sunt viabile; singura monosomie viabilă este cea a cromozomului X (45, X)1;2.
Deoarece trisomiile şi monosomiile sunt în general incompatibile cu viaţa s-a presupus că acestea vor conduce inevitabil la avort spontan. Cu toate acestea, analizele moleculare au arătat că un mic procent al produşilor de concepţie aneuploizi poate fi “salvat” şi da naştere la un făt viabil. In cazul monosomiei “salvarea“ este realizată prin duplicarea singurului cromozom existent, rezultând o pereche de cromozomi cu izodisomie uniparentală (două copii ale aceluiaşi cromozom moştenit de la un părinte). Acest mecanism este exemplificat de o serie de comunicări din literatură în care un copil afectat de fibroză chistică era homozigot pentru mutaţia ΔF508, deşi analiza moleculară a ambilor părinţi a arătat că doar unul dintre aceştia era purtător al mutaţiei. Explicaţia a fost că produsul de concepţie a prezentat o monosomie 7 ce fost “salvată” prin duplicarea cromozomului existent care, întâmplător, era purtător al mutaţiei pentru fibroza chistică. O situaţie similară este posibilă în cazul trisomiei. Cele mai multe trisomii nu sunt viabile, dar dacă unul din cei trei cromozomi este eliminat, rezultă o disomie pentru acea pereche de cromozomi, cu următoarele posibilităţi:
In această situaţie este important momentul în care survine eroarea de diviziune. Astfel, o nondisjuncţie apărută în prima diviziune meiotică va genera o heterodisomie uniparentală (2 cromozomi omologi heterozigoţi proveniţi de la acelaşi părinte), în timp ce o nondisjuncţie în cea de a doua diviziune va rezulta într-o izodisomie uniparentală (două copii ale unui singur cromozom) – vezi figura 19.1.2.; dar crossing-over poate complica situaţia.
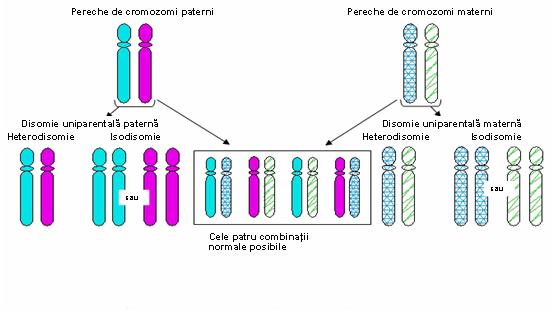
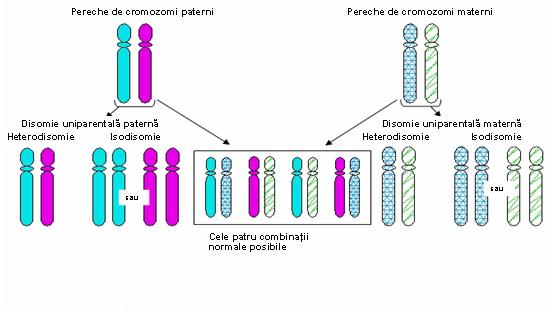
Fig. 19.1.2 Tipuri de disomie uniparentală produse de erori de diviziune
(Adaptare după Uniparental Disomy; Gene Reviews www.be-md.ncbi.nlm.nih.gov)
Deşi se credea că a avea o pereche de cromozomi este suficient, datele actuale arată că, în unele cazuri, este necesar ca perechile să conţină un cromozom de provenienţă maternă şi unul paternă. Problemele de disomie uniparentală vs biparentală ca şi de isodisomie vs heterodisomie au o importanţă majoră pentru fenomenul de amprentare (exprimarea diferenţiată a genelor în funcţie de originea lor parentală)1.
Aneuploidii autozomale
Cea mai obişnuită cauză de retard mental, având o incidenţă de 1 la 700 naşteri, este trisomia 21 sau sindromul Down. Nou-născutul cu trisomie 21 are lungime şi greutate mai mică decât parametri vârstei gestaţionale, prezintă hipotonie musculară, hiperextensibilitate şi reflexe comportamentale (de exemplu refexul Moro) reduse. Capul este brahicefalic, cu occiput turtit şi fontanele largi. Faţa este rotundă, plată şi prezintă o dismorfie sugestivă: epicantus (un repliu in unghiul intern al ochiului), fantele palpebrale oblice în sus şi în afară; nasul mic cu rădăcina turtită şi nările mici şi anteversate; gura deschisă şi protruzie linguală (datorită cavităţii orale mici); urechile mai jos situate, mici şi displazice. Gâtul este scurt, cu exces de piele pe ceafă; mâinile sunt scurte şi late, cu brahidactilie, clinodactilie (incurbare) a degetului V şi, frecvent, un singur pliu de flexie palmară (pliu simian); inconstant sunt prezente unele malformaţii viscerale (defecte cardiace, atrezie duodenală, imperforaţie anală).
Aproximativ 92.5% din persoanele cu sindrom Down prezintă 47 cromozomi (sunt incluse 3 copii ale cromozomului 21) ca urmare a unei nondisjuncţii apărute in meioza. Sub 3% dintre pacienţi exprimă un fenotip mai puţin sever datorită unui mozaicism caracterizat prin 2 linii celulare (47,XX,+21/46, XX sau 47,XY,+21/46,XY) şi aproximativ 5% au doar 46 cromozomi, deoarece cromozomul 21 in exces face parte dintr-o translocaţie robertsoniană sau un alt tip de translocaţie. Faptul că un copil prezintă o translocaţie reprezintă un indiciu că unul dintre părinţi ar putea fi purtător al unei astfel de anomalii structurale şi de aceea este important să se stabilească cariotipul fiecărui părinte pentru a vedea dacă respectivul cuplu prezintă riscul de a mai avea un copil cu sindrom Down la sarcinile ulterioare. Deşi in majoritatea cazurilor un pacient cu sindrom Down deţine 3 copii complete ale cromozomului 21, studiile moleculare ale persoanelor cu rearanjări ale cromozomului 21 au arătat în mod clar că sunt necesare doar 3 copii ale regiunii cromozomului 21 ce conţine benzile 21q22.12-21q22.3, denumită regiunea critică pentru sindromul Down.
Celelalte două trisomii viabile sunt trisomia 13 şi trisomia 18. Trisomia 13 sau sindromul Patau este întâlnită cu o incidenţă de 1:4000-1:10 000 naşteri. Se caracterizează prin microcefalie, defecte ale scalpului, fisură labio-palatină, ciclopie, cataractă, displazie retiniană, polidactilie, pliu simian, călcâi proeminent, defect septal ventricular, polichistoza renală, uter bifid, criptorhidrie, hipospadias, retard mintal sever. Trisomia 18 sau sindromul Edwards, are o incidenţă de 1:8000 nou-născuţi; trăsăturile clinice includ greutate mică la naştere, microcefalie, occiput proeminent, micrognatie, urechi jos inserate cu pavilionul modificat, fisură de palat sau palat ogival, anomalii ale dermatoglifelor, haluce in dosiflexie, defect septal ventricular, rinichi in potcoavă, retard mintal sever.
Dacă pacienţii cu sindrom Down pot supravieţui până în cea de-a doua sau a treia decadă de viaţa, cei cu trisomie 13 sau 18 decedează de obicei în prima lună. Din aceste motive, consilierea genetică include opţiunea de întrerupere a sarcinii.
Aneuploidiile cromozomilor sexuali
Sunt relativ frecvente în populaţie (prevalenţa globala de 1:500 naşteri) şi exprimă un fenotip mai puţin sever în comparaţie cu aneuploidiile autozomale datorită efectului de inactivare a cromozomului X precum şi a numărului limitat de gene situate pe cromozomul Y.
In majoritatea cazurilor, aneuploidiile cromozomilor sexuali sunt reprezentate de patru afecţiuni mai frecvente: trei trisomii şi o monosomie.
Cariotipurile aberante ale persoanelor care prezintă 3 cromozomi X (femei 47,XXX) sau 1 cromozom X şi doi cromozomi Y (bărbaţi 47,XYY) trec adesea neobservate în cursul vieţii. Incidenţa acestora la naştere este de 1:1000, insă, cu excepţia unei staturi mai inalte, nu există alte trasături care să sugereze o anomalie cromozomială. Unele persoane prezintă tulburări de învăţare şi pot fi astfel identificate prin programele şcolare de screening. Femeile XXX şi bărbaţii XXY sunt depistaţi, de asemenea, în clinicile de infertilitate, deşi anomalia citogenetică nu induce tulburări ale fertilităţii. Indivizii XYY au incidenţa crescută de tulburări comportamentale, iar date mai vechi, care ulterior au fost infirmate, au asociat anomalia de cariotip cu o rată mai mare a criminalităţii.
Bărbaţii cu sindromul Klinefelter (47,XXY) au tendinţa de a fi mai inalţi şi mai slabi în comparaţie cu persoanele de aceeaşi vârstă din jurul lor. Principalele trăsături clinice sunt: hipogonadismul postpubertal, dezvoltarea sânilor, infertilitate datorată testiculelor mici cu tubi hialinizati şi azoospermie. Există o formă mai atenuată de sindrom Klinefelter asociată cu un oarecare potenţial fertil ca urmare a unui mozaic cromozomial: 47,XXY/46XY; proporţia relativă de celule XY vs celule XXY la zigot în cursul determinării sexului este esenţială pentru fertilitate.
Cea mai frecvent identificată aneuploidie a cromozomilor sexuali asociată cu fenotip feminin este sindromul Turner – 45,X. Aproximativ jumătate din persoanele cu acest sindrom prezintă monosomia clasică 45,X. Singurul cromozom X este de obicei de origine maternă sugerând faptul că nondisjuncţia meiotică paternă constituie cea mai frecventă sursă de eroare. In plus, faţă de 45,X pot exista şi cariotipuri mai complexe (de exemplu, deleţii de cromozom X, prezenţa unui cromozom X inelar sau izocromozom X, mozaicuri cromozomiale: 45,X/46,XX; 45,X/46,XY). Cea mai severă este situaţia în care există cel puţin o linie celulară cu un cromozom Y parţial sau complet, datorită riscului crescut de gonadoblastom.
Fenotipul asociat sindromului Turner este foarte variabil. Persoanele afectate au de obicei o statură mică (<150 cm) cu disgenezie gonadală şi tulburări de învăţare. Alte trăsături comune includ: o plică de piele ce se intinde de la creasta omoplatului pană la zona nucală (gât palmat), torace in formă de scut, inserţie posterioară joasă a părului, cubitus valgus, malformaţii cardiace sau renale.
Hygroma chistică este aspectul ecografic prenatal caracteristic, corespunzător unei mase chistice septate în regiunea nucală fetală; aceasta apare, cel mai probabil, din cauza dezvoltării anormale a limfaticelor mari fetale. Alte semne ecografice majore în sindromul Turner sunt: defecte ale inimii stângi, malformații renale și scheletale, anasarca sau hidrops fetal. Translucența nucală este, de obicei, crescută in trimestrul I de sarcină.
Persoanele cu sindrom Turner prezintă, de obicei, un nivel normal de inteligenţă, deşi acesta poate fi variabil, existând chiar cazuri de handicapuri mentale semnificative. Deşi infertilitatea este o trăsătura definitorie a acestui sindrom, dezvoltarea tehnicilor de reproducere asistată a permis ca unele femei afectate (in special cele cu cariotipuri mozaicate) să obţină şi să ducă la termen o sarcină normală1;3.
Anomalii cromozomiale structurale
Cromozomii nu sunt structuri statice, astfel că atât în cursul meiozei cât şi al mitozei au loc fenomene de recombinare. Recombinarea este un proces natural care asigură variabilitatea speciilor. Cu toate că există un sistem de reglare foarte dezvoltat pentru a preveni erorile de recombinare, acestea se produc totuşi, conducând uneori la rearanjări care schimbă structura unuia sau mai multor cromozomi. Rearanjările pot fi:
Rearanjările echilibrate sunt de obicei benigne, din punct de vedere clinic, şi tind să se transmită stabil; pot creşte însă riscul de erori survenite în meioză ce pot genera anomalii cromozomiale la făt şi nou-născut. Rearanjările neechilibrate sunt în general asociate cu un fenotip anormal ce include adesea întârziere în dezvoltare şi retard mintal.
Anomaliile cromozomiale structurale sunt reprezentate de:
1. Deleţie – pierderea unei porţiuni dintr-un cromozom ce conduce la monosomie parţială; deleţiile variază în dimensiuni (de la foarte mici la largi) şi pot fi terminale (pierderea cromozomială include şi telomerul) sau interstiţiale (pierderea unei porţiuni interne a cromozomului); deleţiile pot apărea ca urmare a unei rupturi cromozomiale, a unui crossing-over inegal sau a nondisjuncţiilor (vezi figura 19.1.3).
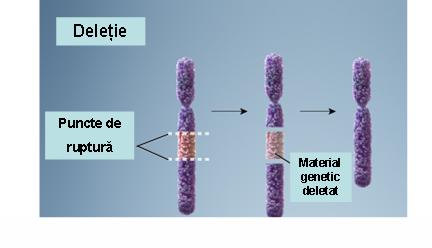
Fig. 19.1.3 Deleţie
(Adaptare după Genetic Homes Reference – Chromosomal Deletion)
2. Duplicaţie – prezenţa unei copii suplimentare a unui segment cromozomial ce conduce la o trisomie parţială; acestă anomalie poate fi de asemenea terminală sau interstiţială; se produc printr-un mecanism similar deleţiilor şi cu cât segmentul duplicat este mai mare cu atât anomaliile clinice vor fi mai severe (vezi figura 19.1.4).
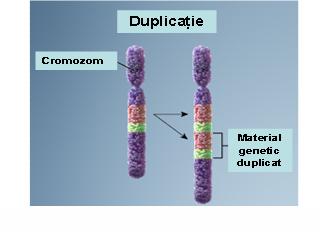
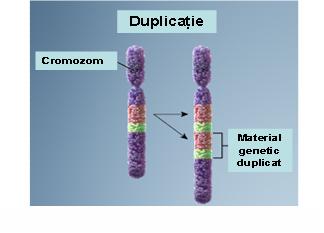
Fig. 19.1.4 Duplicaţie
(Adaptare după Genetic Homes Reference – Chromosomal Duplication)
3. Inversie – amplasarea unor segmente cromozomiale în poziţii inversate faţă de configuraţia normală a genelor; necesită cel puţin două puncte de ruptură la nivelul cromozomului; inversiile pot fi paracentrice (cele două puncte de ruptură se găsesc de aceeaşi parte a centromerului, pe acelaşi braţ) sau pericentrice (punctele de ruptură se găsesc de o parte şi de alta a centromerului şi implică ambele braţe ale cromozomului); majoritatea inversiilor sunt echilibrate, dar dacă ruptura se produce în interiorul unei gene este perturbată sinteza produsului genei respective şi pot fi generate anomalii clinice; există un risc crescut de erori survenite în cursul meiozei, iar purtătorii de inversii prezintă adesea infertilitate sau pierderi precoce de sarcină ca urmare a unor produşi de concepţie cu dezechilibru cromozomial; astfel, în cazul inversiilor paracentrice pot apărea, ca urmare a recombinarii meiotice, cromozomi acentrici sau dicentrici non-viabili (teoretic, nu practic), iar în cazul inversiilor pericentrice este posibil să fie generaţi gameţi cu duplicaţii şi/sau deleţii (vezi figurile 19.1.5 şi 19.1.6).
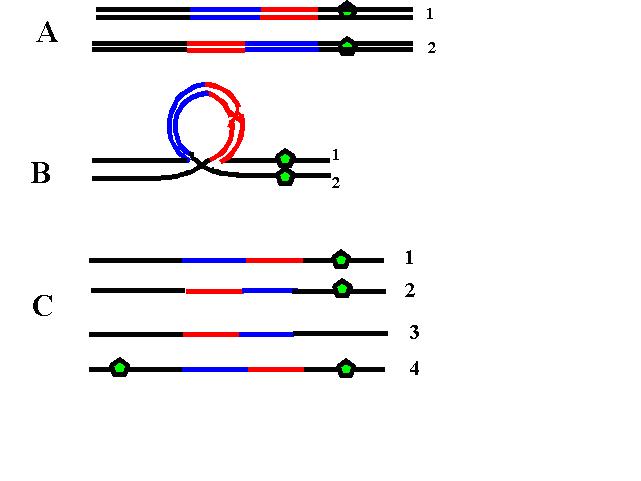
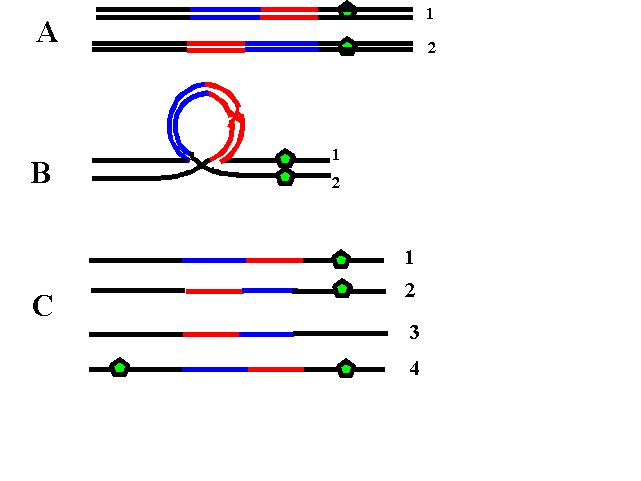
Fig.19.1.5 Inversie paracentrică
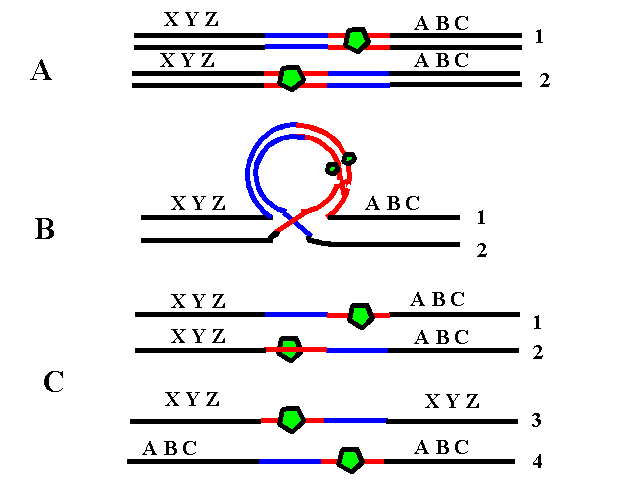
Fig.19.1.6 Inversie pericentrică
4. Translocaţie clasică (reciprocă) – rearanjare care implică unul sau mai mulţi cromozomi neomologi; fiecare cromozom prezintă un singur punct de ruptură, iar segmentele rezultate işi schimbă locul între ele generand doi sau mai mulţi cromozomi derivaţi; la fel ca şi inversiile, majoritatea translocaţiilor sunt echilibrate şi principala problemă o reprezintă riscul crescut de anomalii cromozomiale la produşii de concepţie ai purtătorilor de translocaţii; astfel, în cursul meiozei pot rezulta gameţi cu deleţii/duplicaţii, dacă în anafază cromozomii derivaţi nu segregă împreună (până la o treime din cazuri); cromozomii pot urma şi un model de segregare 3:1 în care 3 cromozomi sunt separaţi unul într-o celulă şi unul în cea de-a doua celulă (vezi figura 19.1.7).
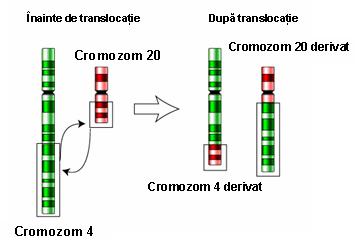
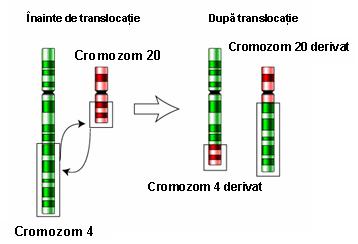
Fig.19.1.7 Translocaţie clasică
(Adaptare după Genetics Primer – http://members.cox.net)
5. Translocaţie robertsoniană – constituie o variantă a translocaţiei clasice; se produce între doi cromozomi acrocentrici prin pierderea, de obicei, a unui segment din braţele scurte, cu păstrarea braţelor lungi; de obicei nu sunt consecinţe clinice deoarece pe toţi cromozomii acrocentrici sunt localizate copii multiple ale genelor ARNr; purtătorii acestui tip de translocaţie au un număr de 45 cromozomi, deoarece doi cromozomi au devenit unul singur din punct de vedere funcţional şi prezintă acelaşi centromer activ; există un risc crescut de nondisjuncţii meiotice rezultând produşi de concepţie cu trisomie faţă de unul din cromozomii rearanjaţi; cel mai obişnuit exemplu este translocaţia ce implică cromozomii 13 şi 14 ai cărei purtători pot da naştere unor copii cu trisomie 13 (cariotip cu 46 cromozomi, 2 copii de cromozom 13 şi translocaţie robertsoniană 13;14); de obicei translocaţia implică doi cromozomi neomologi, însă pot exista situaţii în care sunt afectaţi cromozomi omologi: de exemplu, translocaţia robertsoniană de novo 21;21 ai cărei purtători pot da naştere unor copii cu sindrom Down (vezi figura 19.1.8).
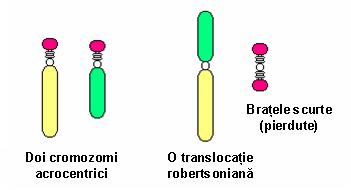
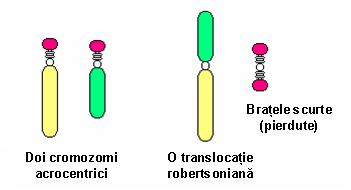
Fig. 19.1.8 Translocaţie robertsoniană
(Adaptare după Cytogenetics – www.ucl.ac.uk)
6. Izocromozom – reprezintă un cromozom ce apare ca urmare a unei erori de diviziune a centromerului: sunt generate două copii ale unui braţ cromozomial cu lipsa celuilalt braţ; rezultă astfel doi cromozomi derivaţi, unul cu o duplicaţie inversată a braţului lung, celălalt cu o duplicaţie inversată a braţului scurt; dacă ambii izocromozomi sunt menţinuţi într-o celulă va rezulta o trisomie pentru cromozomul de bază, condiţie ce este de obicei incompatibilă cu viaţa; dacă însă este menţinut doar un singur izocromozom în celulă va rezulta o trisomie pentru unul din braţele cromozomiale duplicate şi o monosomie pentru celălalt braţ; cel mai cunoscut exemplu este izocromozomul braţului lung al cromozomului X care poate să apară la unele paciente cu sindrom Turner (monosomia pentru braţul scurt al izocromozomului X induce o dezvoltare anormală) (vezi figura 19.1.9).
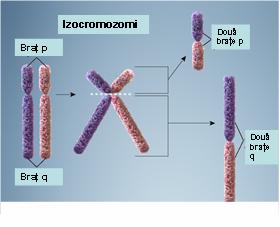
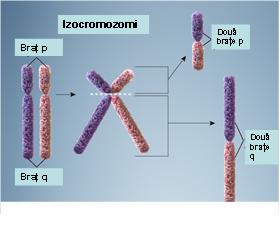
Fig. 19.1.9 Izocromozomi
(Adaptare după Genetic Homes Reference – Isochromosomes)
7. Cromozom inelar – reprezintă un cromozom ce rezultă prin pierderea ambelor telomere şi fuziunea circulară a porţiunii restante în vederea refacerii stabilităţii cromozomiale; în majoritatea cazurilor acest tip de structură este instabil, însă există unele situaţii în care cromozomii inelari pot fi transmişi la celulele-fiice (vezi figura 19.1.10).
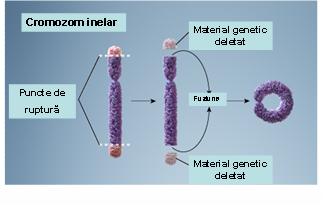
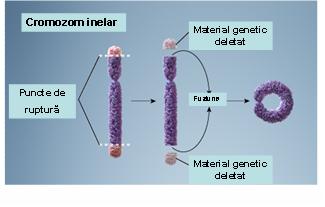
Fig. 19.1.10 Cromozom inelar
(Adaptare după Genetic Homes Reference – Ring Chromosome)
8. Cromozom marker – constituie un cromozom cu un centromer ce este transmis stabil la celulele-fiice, dar care nu poate fi clar identificat, fie datorită dimensiunii sale prea mici, fie datorită aspectului la bandare prea ambiguu. Tehnica SKY (Spectral Karyotyping) constituie un instrument util pentru determinarea originii marker-ilor1;2.
A fost descris un număr semnificativ de sindroame asociate direct cu o anomalie cromozomială structurală.
Sindroamele de deleţii autozomale
Deleţia 4p- (sindromul Wolf-Hirschhorn)
Se caracterizează clinic prin retard de creştere, retard mintal sever, facies în “coif de razboinic grec” cu frunte şi glabela proeminente, sprâncene arcuite şi nas lung, ochi îndepărtaţi (hipertelorism) şi îndreptaţi în jos (fante antimongoliene), pliu cutanat suplimentar în unghiul intern al ochiului (epicantus), strabism, colobomă şi alte anomalii oculare, nas turtit, gura cu buza superioara scurtă, despicătura labială sau palatină (“buză de iepure” sau “gură de lup”), bărbie mică (microretrognaţie), urechi jos implantate, mari, cu forma anormală; surditate, malformaţii cardiace, pulmonare sau renale, hipospadias. Deleţiile braţului scurt al cromozomului 4 pot avea dimensiuni variabile; uneori sunt atât de mici încât nu pot fi detectate decât prin tehnica FISH.
Deleţia 5p- (sindromul Cri-Du-Chat)
Descris în 1963 de către Lejeune şi colaboratorii săi, acest sindrom include un fenotip caractersitic: laringe anormal dezvoltat ce reprezintă cauza ţipătului ascuţit (ca cel al unei pisici), microcefalie, micrognaţie, hipertelorism, fante antimongoliene, pliu palmar unic, hipotonie, retard mintal. Deleţiile pot avea dimensiuni variabile, însă regiunea critică este 5p15.2.
Sindroamele de microdeleţii
Prin definiţie, microdeleţia reprezintă o deleţie foarte mică, care afectează doar o porţiune dintr-o bandă cromozomială. Deşi dimensiunile microdeleţiilor sunt mici, acestea sunt semnificativ mai mari (>500 kb) decât deleţiile moleculare tipice, astfel încât unele pot fi detectate chiar prin efectuarea cariotipului clasic; majoritatea microdeleţiilor sunt însă identificate prin tehnica FISH.
Deşi s-a crezut iniţial că este vorba de deleţii în cadrul unor gene singulare s-a descoperit că multe sindroame se datorează de fapt unor deleţii care cuprind porţiuni din gene adiacente diferite. Mărimea deleţiei şi numărul genelor implicate sunt variabile, astfel că fenotipul exprimat poate să difere semnificativ între persoanele afectate. Aceste sindroame mai sunt denumite sindroame de contiguitate genică:
Sindromul Miller-Dieker
A fost asociat cu o microdeleţie a porţiunii distale a braţului scurt al cromozomului 17 (17p13.3). Elementele clinice caracteristice sunt aspectul particular al feţei şi anomaliile cerebrale: lisencefalia (suprafaţa netedă a emisferelor cerebrale), agenezia de corp calos şi calcificările de pe linia mediană ce explică retardul mintal sever şi convulsiile. Lisencefalia izolată a fost recunoscută ca entitate separată asociată cu o mutaţie a genei LIS1. Sindromul Miller-Dieker este mai complex fiind afectate cel putin două gene.
Sindromul Prader-Willi si sindromul Angelman sunt cele mai cunoscute sindroame de microdeleţie. Ambele prezintă aceeaşi deleţie interstiţială a porţiunii proximale a braţului lung al cromozomului 15 – del(15)(q11.2q13) – însă tabloul clinic este semnificativ diferit. Acest fapt este explicat prin fenomenul de amprentare: dacă deleţia afectează cromozomul de origine paternă rezultă sindromul Prader-Willi, iar dacă este implicat cromozomul de origine maternă se dezvoltă sindromul Angelman.
Pacienţii cu sindrom Prader-Willi prezintă hipotonie severă şi greutate mică la naştere, însă în cursul primului an de viaţă încep să câştige rapid în greutate. Dacă nu se instituie o dietă corespunzătoare se instalează obezitate prin hiperfagie. Alte trăsături clinice includ mâini şi picioare mici, hipogonadism, retard mintal moderat şi un comportament particular cu reacţii violente. Pe de altă parte, pacienţii cu sindrom Angelman prezintă un retard mintal sever, tulburări de vorbire, discursul fiind întrerupt adesea de accese de râs inadecvate situaţiei, hiperactivitate, statură mică, microcefalie, convulsii şi ataxie.
Prin tehnica citogenetică convenţională poate fi detectată deleţia 15q la aproximativ 60% din persoanele cu sindrom Prader-Willi şi doar la 10-20% dintre cele cu sindrom Angelman. Prin tehnica FISH pot fi detectate deleţii în 80-85% din cazuri, pentru ambele sindroame. Faptul că nu pot fi detectate deleţii în toate cazurile a ridicat suspiciunea existenţei unui alt tip de anomalie. La ora actuală se consideră ca sindromul Prader-Willi şi sindromul Angelman sunt asociate cu defecte de amprentare, iar procesele mutaţionale cauzatoare de boală sunt mult mai complexe decât o simplă deleţie a ADN-ului.
Sindromul Williams
A fost asociat cu o deleţie ce cuprinde inclusiv gena elastinei (ELN) localizată în porţiunea proximală a braţului lung a cromozomului 7 (7q11.23). Se caracterizează clinic prin anomalii cardiace, hipertensiune arterială, voce răgusită, îmbătrânirea prematură a pielii, tulburări comportamentale la care se adaugă hipercalcemie şi/sau hipercalciurie. Dacă absenţa elastinei explică multe dintre anomaliile fizice asociate cu acest sindrom, totuşi aceasta nu poate fi asociată cu tulburările comportamentale. In concepţia curentă, sindromul Williams este un sindrom de contiguitate genică, iar variabilitatea fenotipică este dependentă de numărul genelor adiacente afectate de deleţie alături de ELN.
Diagnosticul se stabileşte prin efectuarea cariotipului pentru identificarea rearanjamentelor cromozomiale care pot include regiunea 7q11.23, urmat de testul FISH, cu sonde ADN pentru regiunea critică deletată care include genele ELN şi LIMK1.
Sindromul WAGR
Este un sindrom de contiguitate genică bine definit, ce include tumora Wilms, aniridie, defecte genito-urinare şi retard mintal. Cu excepţia defectelor genito-urinare care par să fie cauzate de o variantă a mutaţiei de la nivelul locusului tumorii Wilms, fiecare din cele 3 anomalii este asociată cu o anumită genă; aceste trei gene sunt dispuse în tandem pe braţul scurt al cromozomului 11. Fenotipul variază în funcţie de mărimea deleţiei. Statistic, 1:3 din copiii cu aniridie vor dezvolta tumora Wilms, în timp ce doar 1:50 din copiii cu tumora Wilms asociază aniridie.
Sindromul deleţiei 22q11.2 ( Sindromul Shprintzen) include: Sindromul DiGeorge si Sindromul Velo-Cardio-Facial
Reprezintă probabil cel mai frecvent sindrom cu microdeleţii, dar adesea este dificil de identificat datorită spectrului larg de manifestari clinice. Acest spectru include hipoplazia sau aplazia timusului (deficite imune) şi a glandelor paratiroide (hipocalcemie), anomalii cardiace, dismorfie facială sugestivă, despicatură palatină sau insuficienţă velo-palatină, anomalii reno-urinare, anomalii scheletice, întârziere în dezvoltarea psihomotorie şi tulburări psihice, dificultăţi de învăţare şi altele. Până la 15% din copii au un părinte afectat de aceeaşi deleţie, dar cu un fenotip mult mai puţin sever. Se pare că deleţia de 3Mb din porţiunea proximală a braţului lung al cromozomului 22 (22q11.2q11.2) se datorează recombinării ce are loc între secvenţele repetitive ce flancheaza regiunea ce este în mod obişnuit deletată. Aceste recombinari intracromozomiale vor genera o structură în formă de buclă ce va fi detectată ca un defect de către enzimele de reparare. Enzimele vor exciza ADN-ul din buclă şi vor reface porţiunea lineară a cromozomului; va rezulta astfel o deleţie cu pierderea tuturor genelor asociate. Deleţia 22q11 este de obicei prea mică pentru a fi identificată prin examenul citogenetic de rutina însă poate fi demonstrată prin tehnica FISH1;3.
Recomandări pentru determinarea cariotipului constituţional
Specimen recoltat – sânge venos4.
Recipient de recoltare – vacutainer ce conţine heparină ca anticoagulant4.
Cantitate recoltată – 5 mL sânge4.
Cauze de respingere a probei – folosirea unui alt tip de anticoagulant; probe vechi, coagulate, hemolizate sau contaminate bacterian4.
Stabilitate probă – maxim 72 ore din momentul recoltării probei până la intrarea acesteia în lucru4.
Metoda – tehnica clasică de bandare G; se va suplimenta cu testarea FISH în funcţie de suspiciunea clinică; sunt analizate 15 metafaze dintre care 5 sunt cariotipizate4.
Raportarea şi interpretarea rezultatelor
In vederea raportării rezultatelor se va folosi următoarea nomenclatură pentru cariotip (în conformitate cu ISCN = International System of Cytogenetic Nomenclature):
– precizarea numărului total de cromozomi;
– precizarea cromozomilor sexuali;
– precizarea oricărei anomalii cromozomiale.
Cele 3 tipuri de date sunt separate prin virgule.
Exemple:
• Femeie cu cariotip normal: 46,XX;
• Bărbat cu cariotip normal: 46,XY;
• Dacă sunt prezente două sau mai multe linii celulare (mosaicism sau chimerism) acestea vor fi indicate secvenţial şi separate prin semnul / iar clona diploidă normală (dacă există) va fi menţionată ultima: 45,X/46,XX; alte clone vor fi prezentate în ordinea dimensiunii acestora, clona cea mai mare fiind prima menţionată – numărul de celule din fiecare clonă va fi indicat de un număr trecut în paranteză: 45,X(15)/47,XXX(3)/46,XX(12);
• In cazul anomaliilor numerice autozomale, numărul total de cromozomi va fi crescut sau redus pentru a indica modificarea globală, iar cromozomul specific dobândit sau pierdut va fi precizat la sfârşit: 47,XX,+13 (trisomie 13 la o persoană de sex feminin); 45,XY,-8 (monosomie 8 la o persoană de sex masculin);
• In cazul anomaliilor numerice constituţionale ale cromozomilor sexuali, nu vor mai fi folosite semnele + sau -: 45,X; 47,XXX; 47,XXY, 47,XYY; pe de altă parte dacă anomaliile numerice ale cromozomilor sexuali sunt dobândite în unele linii celulare maligne vor fi folosite semnele + sau -: 45,X,-Y (o persoană de sex masculin care a pierdut cromozomul Y ca rezultat al unei afecţiuni maligne);
• Dacă sunt prezente, anomaliile cromozomiale structurale vor fi indicate la sfârşitul nomenclaturii pentru a clarifica statusul cromozomului rearanjat; vor fi folosite abrevieri ale anomaliei urmate de cromozomul implicat şi de punctele de ruptură corespunzatoare:
– 46,XX,del(4)(p15): deleţia terminală a braţului scurt al cromozomului 4, la banda 15;
– 46,XX,dup(11)(q13q23): duplicaţia interstiţială a braţului lung al cromozomului 11;
– 46,XY,t(4;9)(q21.2;p22): translocaţie între braţul lung al cromozomului 4 şi braţul scurt al cromozomului 9;
– 46,XY,inv(9)(p11q21.1): inversie pericentrică la nivelul cromozomului 9 între benzile p11 şi q21.1.
La buletinul final va fi ataşată şi cariograma pacientului respectiv1;4.
Bibliografie
1. Constance K. Stein. Applications of Cytogenetics in Modern Pathology. In Henry’s Clinical Diagnosis and Management by Laboratory Methods- Sauders Elsevier 21-Ed 2007, 1267-1281.
2. Chromosome Abnormality. www.web-books.com. Reference Type: Internet Communication.
3. Eberhard Passarge. Karyotype-Phenotype Relationship. In Color Atlas of Genetics, Georg Thieme Verlag KG, 3rd Edition, 2007, 412-416.
4. Laborator Synevo. Referinţele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.