




Informatii generale
Hipercolesterolemia familiala este o afectiune genetica care asociaza nivele extrem de ridicate de LDL – colesterol. Boala se poate transmite:
– autozomal dominant (majoritar), mutatiile ce determina aceasta forma fiind identificate in genele care codifica:
– receptorul LDL (LDLR);
– ligandul receptorului LDL – apoB;
– un modulator al endocitozei hepatice: PCSK9 (proproteina convertaza subtilisin/kexin tip 9);
–autozomal recesiv, mutatiile cauzatoare fiind evidentiate in gena LDLRAP1 ce codifica proteina adaptoare 1 a receptorului LDL necesara internalizarii mediata de clatrina a receptorilor LDL in hepatocite1.
Cele mai multe (60–75%) cazuri de hipercolesterolemie familiala (HF) se datoreaza mutatiilor genei LDLR transmise autozomal dominant, cu un efect doza-gena. Forma heterozigota are o prevalenta de aproximativ 1:500 de persoane, iar cea homozigota de 1:1000000 in intreaga lume. Excesul seric de LDL se depoziteaza in tot organismul si determina aparitia de xantoame, accidente vasculare cerebrale, ateroscleroza si boli cardiovasculare cu debut precoce. Deoarece se asociaza cu un risc crescut pentru boala coronariana prematura, diagnosticul precoce si managementul agresiv de reducere a nivelului de LDL-colesterol ajuta la prevenirea sau incetinirea progresiei aterosclerozei coronariene5.
HF indusa de mutatiile LDRL este o tulburare determinata de absenta sau functionarea necorespunzatoare a receptorului LDL. Gena receptorului LDL este situata pe bratul scurt al cromozomului 19 si cuprinde 18 exoni. Sudhof et al. au constatat ca 13 din cei 18 exoni ai genei ce codifica receptorul LDL au secvente omoloage cu alte gene care codifica proteine diferite. Astfel, 5 exoni codifica o secventa similara cu componenta C9 a complementului; 3 codifica o secventa similara cu o regiune repetitiva a precursorului factorului de crestere epidermala – EGF (engl. epidermal growth factor) si a altor 3 proteine plasmatice ale sistemului de coagulare – factorul IX, factorul X si proteina C, iar 5 exoni codifica secvente nonrepetitive comune cu precursorul EGF.
Receptorul LDL este o proteina transmembranara de 839 aminoacizi, caracterizata prin urmatoarele domenii:
-domeniul de legare (aproximativ 292 aminoacizi, codificat de exonii 2-6);
-domeniu similar cu EGF (aproximativ 400 aminoacizi, codificat de exonii 7-14);
-domeniul bogat in carbohidrati (aproximativ 58 aminoacizi, codificat de exonul 15);
-domeniul transmembranar (circa 22 aminoacizi, codificat de exonul 16);
-domeniul citoplasmatic (aproximativ 50 aminoacizi).
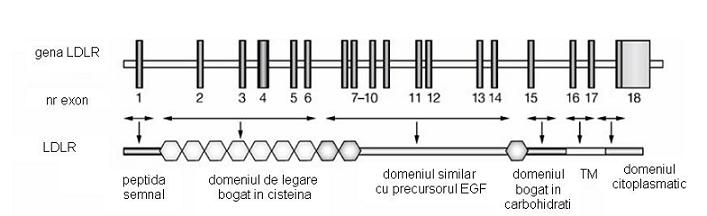
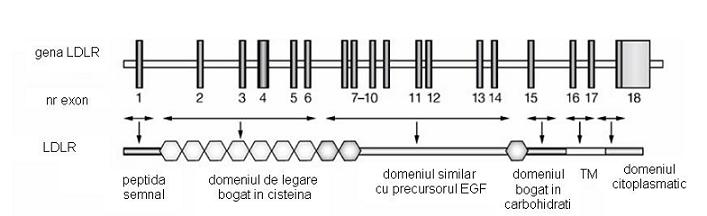
Fig. 1. Modul de organizare al genei ce codifica LDLR
Adaptare dupa Mechanisms of Disease: genetic causes of familial hypercholesterolemia, Anne K Soutar and Rossi P Naoumova, Nature Clinical Practice Cardiovascular Medicine (2007) 4, 214-225
Proteina este sintetizata la nivelul reticulului endoplasmatic rugos sub forma unui precursor de 120 kDa, se maturizeaza in aparatul Golgi ajungand la 160 kDa si este apoi transportata la nivelul membranei celulare prin intermediul veziculelor acoperite de clatrina.
Goldstein si Brown au fost primii care au descris receptorul LDL si tot ei au stabilit ca HF este cauzata de o mutatie autosomal dominanta. De atunci mai mult de 700 mutatii cu impact semnificativ asupra functiei receptorului au fost identificate. Majoritatea se localizeaza la nivelul domeniului care codifica regiunea de legare a receptorului sau in domeniul omolog cu factorul de crestere epidermal (EGF), in regiunea 5′ a genei. Cele mai multe mutatii genice sunt reprezentate de insertii, deletii sau mutatii punctiforme si mai rar (aproximativ 10-15% cazuri) aceste mutatii se datoreaza unor rearanjari mari, cum ar fi deletiile exonice sau duplicatiile5.
Cinci clase de mutatii au fost definite dupa cum urmeaza:
-Clasa 1 cuprinde alelele nule care au ca rezultat absenta completa a receptorilor LDL.
-Clasa 2 cuprinde mutatii ale alelor ce sunt implicate in transportul receptorilor (determina afectarea modului de pliere a receptorului si cauzeaza esecul transportului la suprafata celulelor sau transportul se realizeaza cu succes, dar receptoriii prezinta mutatii).
-Clasa 2a: mutatiile blocheaza complet transportul receptorilor de la reticulul endoplasmatic la aparatul Golgi.
-Clasa 2b: mutatiile blocheaza partial transportul de la reticulul endoplasmatic la complexul Golgi.
-Clasa 3 cuprinde alelele deficiente ce afecteaza legarea receptorului la ligand (LDL si, in unele cazuri, VLDL).
-Clasa 4 cuprinde alelele deficiente ce afecteaza internalizarea receptorilor in vezicule acoperite de clatrina la nivelul hepatocitelor.
-Clasa 5 cuprinde alelele deficiente care impiedica disocierea receptorului de ligand si astfel reciclarea receptorului2.
In hipercolesterolemia familiala functia receptorilor LDL variaza de la absenta totala la aproximativ 25% din activitatea receptorilor normali.
Circa 70% din LDL circulant este captat de ficat prin intermediul receptorului LDL si apoi metabolizat. Capatul amino-terminal al receptorului LDL este situat extracelular si contine in situsul de legare 7 domenii repetitive formate din aproximativ 40 aminoacizi (cisteina este abundenta), cu rol in medierea atasarii lipoproteinelor. Doi liganzi, apolipoproteina B-100 (apoB-100) si apoE (se gaseste in VLDL, IDL, HDL si chilomicroni), se leaga de receptorul LDL si determina activarea acestuia. Interactiunea se realizeaza in prezenta calciului si implica existenta unor aminoacizi incarcati negativ. Domeniul repetitiv 5 este esential pentru legarea apoE, in timp ce regiunile 2-7 sunt necesare pentru atasarea unor liganzi mai mari, cum este apoB1.
Un sindrom aproape imposibil de diferentiat de forma heterozigota a hipercolesterolemiei familiale este determinat de aparitia unor mutatii la nivelul genei care codifica apolipoproteina B-100. Aceasta forma – deficienta ligandului apo-B (FDBL) reprezinta aproximativ 15% din cazurile de HF, se transmite autozomal dominant si are o penetranta de aproape 100%5.
Apolipoproteina B este componenta principala a chilomicronilor si a lipoproteinelor cu densitate mica (LDL). Apare in plasma sub 2 forme, apo B48 si apo B100, prima fiind sintetizata exclusiv la nivelul intestinului, iar a doua de catre ficat. Apo B100 este o proteina mare de 550 kDa (4536 aminoacizi) codificata de o gena situata pe bratul scurt al cromozomului 2 (2p24-p23) si compusa din 26 exoni. Trei mutatii in exonul 26 sunt asociate cu acest tip de hipercolesterolemie. Marea majoritate a cazurilor este cauzata de mutatia la nivelul codonului care codifica aminoacidul 3500 (R3500Q), ce determina substitutia glutaminei cu arginina. Aceasta mutatie determina aparitia unor modificari conformationale la nivelul domeniului de legare la receptor, iar lipoproteinele cu acest tip de apoB au afinitate scazuta (doar 10%) pentru receptorii LDL. Desi numarul si functia receptorilor LDL sunt normale, LDL este preluat ineficient, fapt ce determina aparitia unor nivele serice crescute de LDL-colesterol. Mutatia are o frecventa estimata la 1:500 indivizi in Europa. Cea de a doua mutatie, ce consta in substitutia argininei cu cisteina in pozitia 3531 (R3531C), a fost diagnosticata la doua familii cu origini etnice diferite. Aceasta mutatie produce un defect de afinitate la nivelul receptorilor, dar care determina o scadere cu 30% mai mica decat cea observata la mutatia din pozitia 3500. O a treia mutatie, mai putin frecventa, ce apare la nivelul aceluiasi codon consta in substitutia triptofanului cu arginina (R3500W) si este mai frecventa in Asia, dar a fost identificata si in randul populatiei scotiene.
Prevalenta acestor mutatii difera in randul populatiei, majoritatea cazurilor gasindu-se in populatiile caucaziene. De fapt, cea mai mare incidenta a acestui defect se afla in Europa Centrala si Elvetia (1:200 adulti), iar frecventa scade treptat in bazinul mediteranean si in populatiile europene nordice. In Germania, Marea Britanie si SUA prevalenta variaza intre 1:500 si 1:700 adulti4;5.
Manifestarile clinice ale acestei forme de hipercolesterolemie asociate cu mutatii la nivelul genei care codifica apoB (FDBL) sunt explicate prin acumularea serica de LDL. Acesti pacienti prezinta un risc crescut de aparitie precoce a bolilor cardiovasculare si leziunilor cutanate similare cu cele ale pacientiilor cu forma heterozigota de HF, determinata de mutatii in gena care codifica LDLR. Deoarece receptorii LDL functioneaza normal, concentratiile plasmatice de VLDL, IDL se gasesc in intervalul de referinta.
Nivelele serice crescute de colesterol determina endocitarea acestuia de catre monocite si macrofage, care se vor transforma in celule spumoase si vor contribui la formarea placilor ateromatoase la nivelul endoteliului, cu aparitia prematura a bolilor cardiovasculare. De asemenea colesterolul se acumuleaza in special la nivelul pielii, formand xantelasme si xantoame.
Pacientii cu forma homozigota a HF prezinta xantoame cutanate la nastere sau precoce in copilarie. Mai multe tipuri de xantoame sunt de obicei evidente in primul deceniu de viata si acestea includ:
– xantoame plane (maini, coate, fese sau genunchi), care sunt elemente de diagnostic pentru forma homozigota si se caracterizeaza prin culoarea lor galben-portocalie;
– xantoame tuberculate (maini, coate sau genunchi);
– xantoamele tendoanelor (in special pe tendoanele extensorilor mainilor sau tendonului lui Achile), care apar mai tarziu2.
Copiii cu forma homozigota de boala pot prezenta simptome de cardiopatie ischemica, boli vasculare periferice, boli cerebrovasculare sau stenoza aortica. De asemenea pacientii pot avea simptome articulare, cum ar fi tendinita sau artralgii, istoric de leziuni neobisnuite ale pielii sau arc corneean, care uneori poate fi circumferential2.
Cei mai multi adulti nu supravietuiesc dupa varsta de 30 ani, cu exceptia cazurilor tratate prin metode extreme, cum ar fi transplantul de ficat, afereza LDL sau by-pass ileal, care determina o scadere dramatica a nivelului LDL-colesterolului. Istoricul familial al acestor pacienti prezinta numeroase cazuri de hipercolesterolemie severa si afectiuni cardiace premature2.
Copiii cu forma heterozigota nu prezinta simptome cardiovasculare, dar au un parinte cu hipercolesterolemie severa si, probabil, antecedente personale sau familiale de afectiuni cardiace dezvolate prematur. Statistic, deoarece gena pentru HF se transmite autozomal dominant, jumatate din fratii acestui pacient vor avea de asemenea forma heterozigota a bolii. Dupa varsta de 30 ani mai mult de 60% dintre pacientii netratati cu HF dezvolta xantoame tendinoase, de obicei la nivelul tendonului lui Ahile si tendoanelor extensoare metacarpiene sau interfalangiene de la maini. Majoritatea manualelor sugereaza ca xantoamele de la nivelul tendoanelor la pacientii heterozigoti sunt evidente la inspectie, dar, din pacate, acest lucru nu este vizibil in cele mai multe cazuri. Atenta palpare, mai degraba decat inspectia simpla, poate fi necesara pentru detectarea acestora la nivelul tendonului lui Ahile sau a articulatiilor metacarpofalangiene. Un tendon difuz ingrosat sau cu neregularitati discrete este sugestiv pentru xantoame2.
Adultii cu forma heterozigota au o istorie de indelungata de hipercolesterolemie grava datand din copilarie. Chiar daca pana in prezent pacientul nu a suferit niciun eveniment coronarian acut, simptomele bolii cardiace ischemice sunt frecvente, mai ales daca se asociaza si alti factori de risc cardiovascular (in special fumatul). Bolnavii prezinta de asemenea simptome de tendinita recurenta si artrita. Bolile cardiovasculare precoce si hipercolesterolemia severa sunt prezente la una sau mai multe rude de gradul intai2.
In general heterozigotii HF au cresteri plasmatice de 2 ori mai mari fata de valorile de referinta si dezvolta ateroscleroza coronariana dupa 30 ani. Persoanele homozigote au valori mari ale colesterolului, in general >650 mg/dL, cu prezenta xantoamelor cutanate inainte de 4 ani, bolii coronariene in copilarie si moarte prin infarct miocardic inainte de 20 ani.
Aproximativ 40% din barbatii si 20% din femeile cu o mutatie apoB vor dezvolta boli coronariene. In comparatie cu FH persoanele cu FDBL au manifestari mai putin severe5. Pentru purtatorii acestui tip de mutatie hipercolesterolemia este o caracteristica generala, dar exista un grad ridicat de variabilitate in ceea ce priveste varsta de debut si severitatea simptomelor clinice, desi intr-o familie exista un grad mai mare de similitudine, sugerand ca factorii genetici si de mediu sunt de asemenea implicati in dezvoltarea bolilor cardiovasculare in aceste familii6.
Diagnosticul hipercolesterolemiei, atat forma homozigota cat si cea heterozigota, se bazeaza in principal pe constatarea cresterii severe a nivelului seric de LDL-colesterol in absenta cauzelor secundare de hipercolesterolemie, cu valori ale trigliceridelor cuprinse in intervalul de referinta sau usor crescute. Un diagnostic probabil al formei heterozigote a HF se poate face in cazul in care nivelul LDLc este mai mare de 330 mg/dL sau daca exista xantoame tendinoase. Diagnosticul de confirmare insa se stabileste numai prin analiza genetica. Acesta este important pentru depistarea precoce a membrilor de familie afectati, in special a subiectilor tineri.
Diagnosticul molecular al HF conduce la cresterea proportiei de pacienti care incep sau intensifica terapia anticolesterolemianta si, in consecinta, la o scadere adecvata a concentratiei LDL-colesterolului.
Screening-ul bolilor genetice la copii este o problema delicata si trebuie efectuata in conformitate cu reglementarile speciale din diferitele tari. In tarile nordice numai copiii cu varsta mai mare de 16 ani pot participa la programul national de screening genetic. In Norvegia screening-ul copiilor mai mici este posibil dupa obtinerea acordului prealabil al parintilor.
Dupa o evaluare atenta a riscurilor si beneficiilor individuale, s-a stabilit ca numai copiii cu cel mai mare risc de a dezvolta hipercolesterolemie ar trebui luati in considerare pentru tratamentul cu statine6.
Diagnosticul prenatal la gravidele cu risc crescut pentru o mutatie LDLR sau apoB este posibil prin analiza ADN-ul extras din celule fetale obtinute prin amniocenteza, de obicei efectuata la aproximativ 15-18 saptamani de gestatie, sau biopsia vilozitatilor coriale la aproximativ 10-12 saptamani de gestatie. Mutatiile cauzatoare de boala trebuie sa fie identificate inainte de testarea prenatala la un membru afectat al familiei.
Conform recomandarilor actuale diagnosticul prenatal nu este adecvat pentru HF, cu exceptia unei sarcini cu risc de HF homozigota. Exceptand aceste familii nu exista motive pentru intreruperea unei sarcinii cu fat heterozigot HF, deoarece in aceasta tulburare simptomele clinice au debut tardiv, iar hiperlipemia si riscul de aparitie a bolilor cardiovasculare pot fi modificate prin stilul de viata si sunt tratabile prin diverse terapii. Forma homozigota a HF are o prevalenta de 1 la 1000000 nasteri vii, desi in anumite grupuri etnice (in care casatoriile intre rude sunt numeroase) frecventa este mai mare. In astfel de familii identificarea unui copil cu forma homozigota se face de obicei dupa ce acesta a fost deja nascut6.
Recomandari pentru testarea genetica – diagnosticul hipercolesterolemiei familiale (asociata cu mutatii la nivelul genelor care codifica LDLR si apoB-100) la persoanele cu valori crescute ale colesterolului si diferentierea acesteia de alte cauze de hiperlipemie5.
Specimen recoltat – sange venos3.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant3.
Cantitate recoltata – 5 mL sange3.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate3.
Stabilitate proba – 7 zile la 2-8ºC3.
Metoda – sunt disponibile 2 tipuri de teste:
a) pentru mutatiile LDLR: secventiere completa a genei + analiza deletiilor/duplicatiilor MLPA;
b) pentru mutatiile ligandului apo-B: secventiere exon 26 pentru depistarea mutatiilor R3500Q, R3531C, R3500W3.
Raportarea si interpretarea rezultatelor
Vor fi comunicate mutatiile depistate in genele LDLR si/sau apoB-1003.
Bibliografie
1. Anne K Soutar, Rossi P Naoumova. Mechanisms of Disease: genetic causis of familial hypercholesterolemia. In Nature Clinical Practice – Cardiovsacular Medicine, April 2007, Vol.4, Nr.4.
2. Elena Citkowitz. Hypercholesterolemia, Familial www.emedicine.medscape.com, Ref Type: Internet Communication.
3. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
4. Ludivina Robles-Osorio, Ma. Luisa Ordoñez, Carlos A. Aguilar-Salinas, Moisés Aurón-Gómez, Ma. Teresa Tusié-Luna, Francisco J. Gómez-Pérez and Juan A. Rull-Rodrigo Familial Hypercholesterolemia Due to Ligand-Defective Apolipoprotein B100. First Case Report in a Mexican Family. In Archives of Medical Research 34:70–75, 2003.
5. Mayo Clinic/Mayo Medical Laboratories. Test Catalog. Familial Hypercholesterolemia/Autosomal Dominant Hypercholesterolemia Genetic Testing Reflex Panel. www.mayomedicallaboratories.com. Ref Type: Internet Communication
6. S.E. Humphries, D. Galton and P. Nicholls. Genetic testing for familial hypercholesterolaemia: practical and ethical issues, Q J Med 90:169–181, 1997.